Bourneville–Pringle disease with bilateral renal angiomyolipoma and epitheloid monomorphic angiomyolipoma in a 43-year-old woman
DOI: 10.22591/magyurol.2024.3.matek.150
Authors:
Máté Kinga dr., Márványkövi Fanni dr., Vrecenár László dr.,
Zóber Tamás dr., Buzogány István dr., Beöthe Tamás dr.
Péterfy Sándor utcai Kórház, Urológia Osztály, Budapest (osztályvezető: Buzogány István dr.)
Summary
Introduction: The Bourneville Pringle disease, also called Tuberous Sclerosis Complex (TSC) is a rare genetic disorder with dominant autosomal inheritance. It is a rare neurocutaneous disorder, which can affect numerous internal organs, for example the kidney(s), the heart, and the lungs. The formation of multiple hamartomas is causing the multiorgan system involvement.
Case presentation: A 43-year-old patient presents with tuberous sclerosis, an autosomal dominant inherited neurocutaneous disorder which variably affects different organs, among them the kidneys. During follow-up the abdominal Computer Tomography (CT) showed bilateral angiolipomatosis (AML) and a fat-poor mass with a different solid aspect on the left side. A fat-poor AML can be a challenge to diagnose. It is vital to exclude malignancy to decide on further management. A CT guided biopsy was performed. The results showed an atypical renal angiomyolipoma, a rare monomorph, epitheloid type. A genetic examination was performed, which showed a mutation of the TSC2 genes.
Conclusion: The Bourneville Pringle disease is a rare genetic, neurocutaneous disease, which can affect multiple organs by the formation of hamartomas, including renal manifestation as AML. In case of radiological findings of a fat poor CT component AML it is essential to keep in mind the possibility of a rare monomorph epitheloid AML and if there is any doubt, it is crucial to exclude malignancy by using biopsy. A fat-poor AML can be a challenge to diagnose. It is vital to exclude malignancy, to take the size of the kidney AML in consideration, the high risk of potentially massive hemorrhage to decide on further management.
LAPSZÁM: MAGYAR UROLÓGIA | 2024 | 36. ÉVFOLYAM, 3. SZÁM
Összefoglalás
Bevezetés: A Bourneville–Pringle-kór, más néven tuberosus sclerosis komplex (TSC), ritka genetikai rendellenesség, amely domináns autoszómális öröklődéssel jár. Ez egy ritka neurokután rendellenesség, amely számos belső szervet érinthet, például a veséket, a szívet és a tüdőt. A hamartómák kialakulása okozza a többszervű érintettséget.
Esetbemutatás: Egy 43 éves, tuberosus sclerosisban szenvedő beteg esetbemutatása. A tuberosus sclerosis egy autoszomális domináns öröklődéssel járó neurokután rendellenesség, amely változó mértékben érintheti a különböző szerveket, köztük a veséket. A követés során a hasi komputertomográfia (CT) kétoldali angiolipomatózist (AML) és egy zsírszegény elváltozást mutatott a bal vesében. A zsírszegény AML diagnosztizálása kihívást jelenthet. A rosszindulatú elváltozást minden esetben ki kell zárni. CT-vezérelt vesebiopszia történt. Az eredmények atipikus, vesében kialakuló angiomyolipomát, ritka monomorf, epitheloid típusát mutatták ki. Genetikai vizsgálat történt, amely során a TSC2-gének mutációját találtuk.
Következtetés: A Bourneville–Pringle-kór ritka genetikai neurokután betegség, amely több szervet érinthet a hamartómák különböző szervekben való kialakulásával, ideértve a vesékben előforduló AML-t is. A zsírszegény AML diagnosztizálása esetén fontos figyelembe venni a ritka monomorf epitheloid AML lehetőségét, és ha kétség merül fel, elengedhetetlen a rosszindulatú elváltozás kizárása biopsziával. A zsírszegény AML diagnosztizálása kihívást jelenthet. A további kezelés eldöntése során fontos figyelembe venni a potenciálisan súlyos vérzés magas kockázatát.
Bevezetés
A Bourneville–Pringle-kór, más néven Tuberous Sclerosis Complex (TSC), ritka genetikai rendellenesség, amely domináns autoszomális öröklődéssel jár (1). A betegség előfordulása körülbelül 1:12, de ez országonként változhat.
A betegség nevét a francia neurológus, Désiré-Magloire-Bourneville után kapta, aki először 1880-ban írta le. Később Pringle kiegészítette a szindróma leírását a bőr megnyilvánulásaival, konkrétan az arc angiofibromáival, más néven adenoma sebaceummal. Ez a ritka neurokután rendellenesség, amely számos belső szervet érinthet, például a veséket, a szívet és a tüdőt. A hamartómák kialakulása okozza a többszervű érintettséget. A tuberosus sclerosis a TSC1 (hamartin) és/vagy a TSC2 (tuberin) gének mutációja következtében alakul ki (2).
A Bourneville–Pringle-kór diagnosztikai kritériumait 2012-ben felülvizsgálták. A tuberosus sclerosis complex diagnózisához két fő kritérium vagy egy fő kritérium és két mellékkritérium jelenléte szükséges (3). A fő kritériumok közé tartoznak a pigmenthiányos foltok (≥3, legalább 5 mm átmérőjű), angiofibromák (≥3) vagy cápabőrfolt, körömfibroma (≥2), Shagreen-folt, többszörös retinalis hamartómák, agykérgi göbök, subependymalis csomók, subependymalis óriás sejtes astrocytomák, szívritmuszavarok, lymphangioleiomyomatosis (LAM), angiomyolipomák (≥2). A mellékkritériumok közé tartoznak a „konfett” bőrelváltozások, fogzománci árkok, (>3), szájüregi fibromák (≥2), depigmentált foltok a retinán, többszörös veseciszták, nem vese hamartomák.
A hamartomák kialakulása különböző szervekben okozza a tüneteket és felelős bizonyos klinikai jellemzők megnyilvánulásáért (4). A bőr megnyilvánulásai közé tartoznak a facialis angiofibromák (adenoma sebaceum), periungualis angiofibromák, cápabőrfolt, pigmenthiányos foltok, fibrotikus plakkok (5, 6). A neurológiai megnyilvánulások közé tartozik az epilepszia, subependymalis óriás sejtes astrocytomák kialakulása, TSC-hez társuló neuropszichiátriai rendellenességek, értelmi fogyatékosság, autizmus (7). A tüdő szintjén megjelenő elváltozás a lymphangioleiomyomatosis (LAM), amely leggyakrabban nehézlégzést és ritkábban köhögést, mellkasi fájdalmat és hemoptízist okoz. A vese megnyilvánulások közül leggyakrabban az angiomyolipomák és ciszták jelentkeznek, de ritka esetekben a vese sejtes karcinómái, epitheloid monomorf angiomyolipomát és onkocytomát is leírtak (7, 8). Ha érintett a szív, kardiális rhabdomyomákat találhatunk.
Esetismertetés
43 éves nő tuberosus sclerosis complexben, Bourneville–Pringle-kórban szenved, amelyet 29 éves korában diagnosztizáltak. A klinikai tünetek a bőrpanaszokkal kezdődtek. Először az arcán alakultak ki angiofibromák, adenoma sebaceum. Később a periungualis fibromákkal és a hátán megjelent hipopigmentált foltokkal jelentkezett a bőrgyógyászati osztályon (1. ábra).
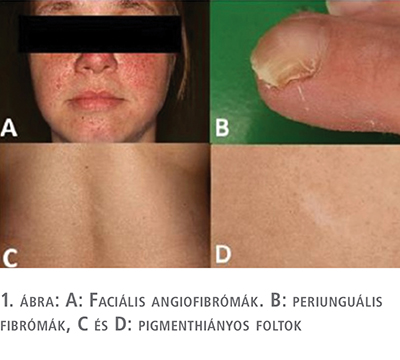
Serdülőkorában epilepsziás rohamai voltak, amelyek a Pregabalin kezelésével megszűntek 2006-ban az hasi ultrahangvizsgálat bilaterálisan megnagyobbodott veséket mutatott, eltérő méretű és echogenitású elváltozásokkal, valamint a bal vese felső pólusán egy inhomogén elváltozást. Hasi CT-vizsgálat történt, amely a fenti veseelváltozásokat lehetséges kétoldali angiolipomának véleményezte. A bal vese felső pólusán található inhomogén képletet zsírszegény AML-ként írták le, amely esetben a malignitás sem volt biztonsággal kizárható. A májelváltozásokat máj angiolipomáknak tartották (2. ábra).
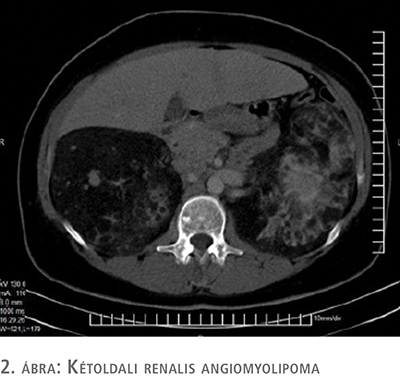
A malignitást megerősíteni vagy kizárni, CT-vezérelt biopsziát végeztek, amelyet 2 patológus vizsgált meg. A szövettani eredmények atipikus vese angiomyolipomát mutattak, ritka monomorf, epitheloid típusút, amely ismert jóindulatú elváltozás, de bizonyos mintázatok potenciálisan rosszindulatúak, és további megfigyelést igényelnek. Ezek a mintázatok a malignizálódás lehetőségét jelzik, és a szövettani eredmények elengedhetetlenek ezen mintázatok meghatározásához. A koponya CT-vizsgálat és az agyi mágneses rezonancia képalkotás (MRI) tipikus elváltozásokat mutatott a tuberosus sclerosisra. A beteget a szemészeti osztályra irányították, ahol hamartomát találtak a szemfenéki vizsgálat során. A genetikai vizsgálat a beteg mintájából mutációt mutatott ki a TSC2-génnél. A tüdő szintjén a lymphangioleiomyomatosis (LAM) diagnosztizálása az év elején történt, köhögéssel és nehézlégzéssel. Az irányelvek szerint a vesében lévő angiomyolipoma esetén, ha az elváltozás nagyobb, mint 4 cm, vagy az aneurizma nagyobb mint 5 mm átmérőjű, fennáll a potenciálisan masszív vérzés magas kockázata (9). A szövettani eredmények és az immuno-hisztokémiai jellemzők alapján a jóindulatú EM-AML-formára utalóan, az irányelveknek megfelelően, figyelembe véve a léziók méretét és a potenciálisan masszív vérzés magas kockázatát, felajánlották a műtét lehetőségét. A beteg az urológiai műtétet visszautasította, és csak a bőrelváltozásai miatt döntött a beavatkozás mellett.
A beteget 14 éve követik szoros kontrollal. A nemzetközi irányelvek legalább évente javasolják a vesefunkció és vérnyomás ellenőrzését, valamint a veseképalkotást 1-3 évente. Az esetünkben a betegnél másodlagos hipertónia alakult ki, amelyet béta-blokkolóval, Metoprolol (Betaloc) 50 mg 2×1 és valsartan plusz hidroklorotiazid (Co-Valsacor) 80/12,5 mg 2×1/2 tabl. kezelnek, és kedvező eredményt mutatnak. Az utolsó vesefunkcióteszt eredményei normáltartományok között voltak: kreatinin 76 U/l, karbamid 4,4 mmol/l, eGFR >60 ml/perc/1,73 m2. A veseképalkotás tekintetében MRI- és CT-vizsgálatokat végeztek, kezdetben félévente, majd évente. A követési időszak alatt végzett veseképalkotás eredményei nem mutattak jelentős eltérést. Az epilepszia hatékonyan kezelhető a Pregabalin (Lyrica) 2×75 mg-mal. 14 éves követés után a beteg stabil állapotban van, remisszióban van a vesére vonatkozó elváltozások terén, veseműtét nélkül. A tüdőmegnyilvánulás (LAM) diagnózisát illetően a beteg az mTOR-gátló, Everolimus terápiára várja az engedélyt.
Eredmények és megbeszélés
A tuberosus sclerosis egy genetikai rendellenesség, amely változatosan érinti az idegrendszert, a bőrt, és más belső szerveket, mint például a veséket, tüdőt és szívet. A klinikai megnyilvánulások a többszervi érintettség következményei. A vesében megjelenő elváltozások leggyakoribb formái a ciszták és az angiomyolipomák. Az AML egy jóindulatú daganat, amelyben zsírszövet, ér és simaizomsejtek találhatóak. A klasszikus AML-t a magas zsírszöveti komponens jellemzi. Az irodalomban néhány ritka esetben előfordul vesesejtes karcinóma, onkocytoma és egy ritka monomorf epitheloid AML (10). A zsírszegény AML kihívást jelent a diagnózis felállításában. A legtöbb esetben a malignitást sem lehet kizárni. A CT-vezérelt biopszia és a szövettani eredmények segíthetnek a diagnózisban és a további kezelési és követési stratégia kiválasztásában. Az immunhisztokémiai vizsgálatok, amelyek a melanin-A és HMB-45 jelenlétét mutatják ki, segíthetnek megkülönböztetni a ritka monomorf, epitheloid típust a vesesejtes karcinómától (11). Az epitheloid AML esetén az epitheloid sejtek dominálnak. Az irodalomban nincsenek pontos adatok arra vonatkozóan, hogy mekkora arányban kell jelen lenni az epitheloid sejteknek ahhoz, hogy egy klasszikus AML-t epitheloid AML-ként definiáljunk. Aydin és munkatársai vizsgálták 194 vese AML esetének klinikopatológiai jellemzőit, kiemelve az epitheloid komponenst, legalább 10% arányt állapítottak meg az epitheloid AML-nek való meghatározásához.
A tuberosus sclerosisban a vese angiomyolipomák gyakori klinikai megnyilvánulások, amelyek veszélyes szövődmények megelőzése érdekében körültekintő megfigyelést igényelnek. Gondosan kiválasztott betegeknél konzervatív kezelés, szelektív embolizáció, ablációs terápiák, mTOR-gátló terápiák és sebészi beavatkozások alkalmazhatóak. Yamakado és munkatársai jelentős összefüggést találtak az AML mérete, az aneurizma mérete, valamint a vérzés kockázata között (12). A mi esetünkben a vese AML méretét, az irányelveket (4 cm-nél nagyobb elváltozás vagy 5 mm-nél nagyobb átmérőjű aneurizma, amely magas vérzési kockázattal jár) figyelembe véve radikális nephrectomiát javasoltunk, de a beteg minden sebészeti beavatkozást visszautasított. Az elváltozás anatómiája nem tette lehetővé a szelektív embolizáció lehetőségét. A beteg jelenleg az mTOR-gátló, Everolimus terápia jóváhagyását várja.
Következtetés
A Bourneville–Pringle-kór ritka genetikai neurokután betegség, amely a hamartomák kialakulása révén számos szervet érinthet, beleértve a veséket is, angiomyolipoma formájában. A zsírszegény CT komponensű AML esetén fontos szem előtt tartani a ritka monomorf epitheloid AML lehetőségét, és ha bármilyen kétség merül fel, elengedhetetlen a malignitás kizárása biopsziával. A zsírmentes AML diagnózisa kihívást jelent. Fontos a malignitás kizárása, ugyanakkor fontos figyelembe venni a vesében lévő AML méretét és a potenciálisan masszív vérzés magas kockázatát, a további kezelés kiválasztásához.
Irodalom
1. Alshaiji JM, Spock CR, Connelly EA, Schachner LA. Facial angiofibromas in a mosaic pattern tuberous sclerosis: A case report. Dermatol Online J 2012; 18: 8. https://doi.org/10.5070/D32005V8W3
2. Rodrigues DA, Gomes CM, Costa IM. Tuberous sclerosis complex. An Bras Dermatol 2012; 87: 184–96.
https://doi.org/10.1590/S0365-05962012000200001
3. Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: Revised clinical diagnostic criteria. J Child Neurol 1998; 13: 624–8. https://doi.org/10.1177/088307389801301206
4. Kwiatkowski DJ. Tuberous sclerosis: From tubers to mTOR. Ann Hum Genet 2003; 67(Pt 1): 87–96.
https://doi.org/10.1046/j.1469-1809.2003.00012.x
5. Webb DW, Clarke A, Fryer A, Osborne JP. The cutaneous features of tuberous sclerosis: A population study. Br J Dermatol 1996; 135: 1–5.
https://doi.org/10.1111/j.1365-2133.1996.tb03597.x
6. Sahar El Aoud, et al. Tuberous Sclerosis Complex (Bourneville-Pringle Disease) in a 25-year- old Female with Bilateral Renal Angiomyolipoma and Secondary Hypertension, Saudi J Kidney Dis Transpl 2017; 28(3): 633–638.
https://doi.org/10.4103/1319-2442.206461
7. Rosser T, Panigrahy A, McClintock W. The diverse clinical manifestations of tuberous sclerosis complex: A review. Semin Pediatr Neurol 2006; 13: 27–36. https://doi.org/10.1016/j.spen.2006.01.008
8. Ben Hamida F, Gorsane I, Gharbi C, et al. Renal manifestations in tuberous sclerosis. Rev Med Interne 2006; 27: 836–42.
https://doi.org/10.1016/j.revmed.2006.07.022
9. Kapoor, et al. Evolving Strategies in the Treatment of Tuberous Sclerosis Complex-associated Angiomyolipomas (TSC-AML) Urology 2016 Mar; 89: 19–26. https://doi.org/10.1016/j.urology.2015.12.009
10. Henske EP, Józ´wiak S, Kingswood JC, et al. Tuberous sclerosis complex. Nature Reviews Disease Primers 2016; 2: 16035.
https://doi.org/10.1038/nrdp.2016.35
11. Bradley P. Dixon, John C. Hulbert, John J. Bissler. Tuberous Sclerosis Complex Renal Disease, Nephron Exp Nephrol 2010 Nov; 118(1): e15–e20. https://doi.org/10.1159/000320891
12. Yamakado K, Tanaka N, Nakagawa T, Kobayashi S, Yanagawa M, Takeda K. Renal angiomyolipoma: relationships between tumor size, aneurysm formation, and rupture. Radiology 2002; 225: 78–82.
https://doi.org/10.1148/radiol.2251011477