Complete androgen insensitivity syndrome – case presentation
DOI: 10.22591/magyurol.2025.2.barkoczia.60
Authors:
Barkóczi Alexandra dr., Dócs János dr., Berczi Csaba dr., Flaskó Tibor dr.
Debreceni Egyetem, Általános Orvostudományi Kar, Urológiai Tanszék, Debrecen (igazgató: Flaskó Tibor dr.)
Summary
Introduction: Complete androgen insensitivity syndrome (CAIS) is an X-related congenital malformation and associated with androgen receptor mutation. We present a CAIS case in our department.
Case presentation: 30 year old female was examined with primary amenorrhoea in 2021. Normal female external genitalia were seen on physical examination. The pelvic MRI showed no uterus and upper vagina, which suggested Mayer–Rokitansky–Küster–Hauser syndrome. Two years later the patient had a vaginal reconstruction surgery. After the operation she visited our department. Chromosome analysis showed 46, XY karyotype and complete androgen insensitivity syndrome. Male hormone levels were elevated. Due to the increased risk of malignant tumour we performed laparoscopic gonadectomy. The final histology showed atrophic testicular tissue, there were no sign of malignant cells. Hormone levels normalised after the procedure. We started oral oestrogen supplementation.
Summary: CAIS is a rare sexual dysgenesis (DSD) syndrome, which is characterized by absent androgen effect. In the absence of testosterone effect, the patient has normal female external genitalia with male gonads. Due to its rarity and the normal visual appearance, CAIS is often misdiagnosed or diagnosed late, after puberty. The early diagnosis could be achieved by the usage of prenatal diagnostic tools and chromosome and hormone tests in the suspicion of DSD.
LAPSZÁM: MAGYAR UROLÓGIA | 2025 | 37. ÉVFOLYAM, 2. SZÁM
Összefoglalás
Bevezetés: A teljes androgén inszenzitivitási szindróma (CAIS) egy X-kromoszómához kötött öröklődésű fejlődési rendellenesség, amelynek hátterében az androgénreceptor mutációja áll. A következőkben egy posztpubertásban felismert CAIS esetét mutatjuk be.
Esetismertetés: 30 éves női fenotípusú beteg kivizsgálása 2021-ben indult primer amenorrhoea miatt. Fizikális vizsgálattal női külső nemi szervek voltak láthatóak. Kismedencei MR-vizsgálat történt, amelyen uterus, felső hüvely szakasz nem került leírásra, mindkét oldalon ovárium volt látható. A látott kép alapján Mayer–Rokitansky–Küster–Hauser-szindróma merült fel. Két évvel később hüvelyi rekonstrukciós műtét történt. A műtétet követően jelentkezett a Debreceni Egyetemen. Kromoszómavizsgálat 46, XY kariotípust, valamint teljes androgén inszenzitivitási szindrómát igazolt. Hormonvizsgálat során emelkedett férfi hormonszintek voltak láthatóak. A magasabb malignitás rizikó miatt a korábban ováriumnak véleményezett gonádok laparoszkópos eltávolítását végeztük. A szövettan atrófiás hereállományt mutatott, malignitás nem volt jelen. A posztoperatív hormonszintek normalizálódtak, per os ösztrogénpótlást indítottunk a női nemi jellegek fenntartása érdekében.
Megbeszélés: A CAIS egy ritka szexuális diszgenezis szindróma (DSD), amely teljes androgénhatástalansággal jellemezhető. Tesztoszteronhatás hiányában női külső genitáliák fejlődnek ki, férfi gonádok mellett. A külsőleg normál megjelenés, valamint a ritka előfordulás miatt gyakran nem, vagy csak késői életszakaszban, pubertást követően kerül felismerésre. A prenatális diagnosztika szélesebb körben való alkalmazása, DSD gyanú esetén a kromoszómaanalízis és hormonvizsgálat segíthetné ezen betegek pontosabb, mielőbbi felismerését.
Bevezetés
A teljes androgén inszenzitivitási szindróma (Complete Androgen Insensitivity Syndrome – CAIS) egy szexuális diszgenezis szindróma (Differences of Sex Development – DSD), amely egy X-kromoszómához kötött öröklődésű fejlődési rendellenesség. Hátterében az Xq11-12 lokalizációban kódolódó androgénreceptor (AR) mutáció áll, amely miatt a 46, XY kariotípusú fiú magzatban tesztoszteronhatás nem alakul ki. Ennek eredményeképpen férfi gonádok mellett női külső genitáliák fejlődnek ki. Gyakorisága 1:20 000 a fiú magzatok között (1), a 46, XY DSD-k közül a leggyakrabban előforduló rendellenesség (1, 2). A külsőleg normál megjelenés, valamint a ritka előfordulás miatt gyakran nem, vagy csak késői életszakaszban, pubertást követően kerül felismerésre. A következőkben egy posztpubertásban felismert CAIS esetét mutatjuk be.
Esetbemutatás
A 30 éves női fenotípusú beteg kivizsgálása primer amenorrhoea miatt indult 2021-ben más intézetben. Nőgyógyászati fizikális vizsgálattal ép külső női nemi szervek voltak láthatóak, az 5-7 cm hosszú, tágulékony hüvely mellett porció nem volt azonosítható. A látott kép alapján felvetették Mayer–Rokitansky–Küster–Hauser-szindróma (MRKH-szindróma) lehetőségét. Ekkor további kivizsgálás, hormon- vagy kromoszómavizsgálat nem történt. Két évvel később történt MR-vizsgálat, amelyen a hüvely felső szakasza, valamint az uterus nem volt látható, valamint a lágyékcsatornában mindkét oldalon ováriumnak megfelelő képlet került leírásra (1–2. ábra).
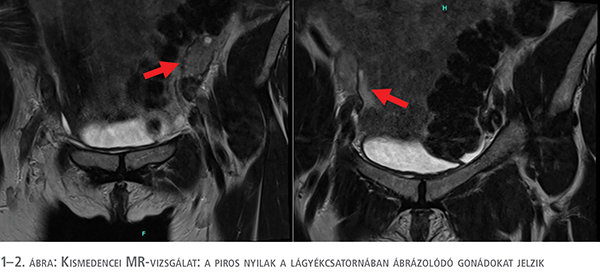
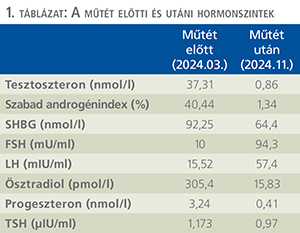
A nemi élet igénye miatt hüvelyrekonstrukciós műtétet javasoltak. Robot-asszisztált Vechietti-féle műtétet végeztek, amely egy minimál invazív neovagina-képző technika. A műtét során egy oliva alakú hüvelyi dilatátort helyeznek be, amelyet subperitoneálisan rögzítenek, majd a posztoperatív szakban 7-10 nap alatt napi 1-2 cm-rel nyújtják a hüvelyt. Később a dilatátort eltávolítják, majd a beteg otthonában további hüvelytágítást végez rendszeresen a későbbi szűkület, illetve rövidülés megelőzése miatt (3). Betegünknél 6 napig alkalmazták az eszköz folyamatos húzását, majd eltávolították. Ezután sebgyógyulási zavar jelentkezett, amely konzervatív kezelés mellett megszűnt. Ezt követően érkezett a Debreceni Egyetem Nőgyógyászati, illetve Urológiai Klinikájára. Endokrinológiai kivizsgálás indult MRKH-szindróma igazolása céljából. A vizsgálatok során emelkedett férfi nemi hormonszintek igazolódtak: tesztoszteron: 37,31 nmol/l, szabad androgén index: 40,44 volt (1. táblázat). Ismételt kismedencei MR-vizsgálat történt, amelyen a korábban leírt ováriumnak imponáló képlet továbbra is a hasfal szintjében volt látható, szabályos megjelenésű uterus helyett egy 15×25×5 mm-es köteges elváltozás került leírásra. Kromoszómaanalízis során 46,XY kariotípus, valamint teljes androgén inszenzitivitás (CAIS) igazolódott. Ezt követően a lágyékcsatorna belső gyűrűjénél található gonádok laparoszkópos eltávolítását végeztük klinikánkon (3. ábra).
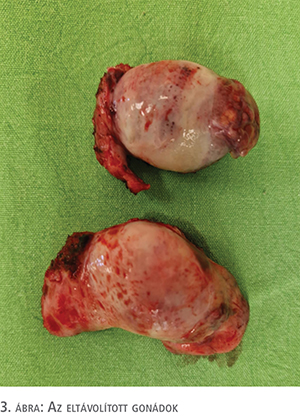
A szövettani vizsgálat primitív atrófiás hereállományt igazolt, Sertoli- és Leydig-sejtes hiperpláziával. Germinális hám vagy ováriumszövet nem volt látható. A műtétet követően történt ismételt hormonvizsgálatban a férfi nemi hormonok szintje lecsökkent (1. táblázat). Hormonpótlás céljából ösztradiol per os adását kezdtük.
Megbeszélés
A CAIS, vagy más néven Morris-szindróma, egy ritka genetikai rendellenesség, amely az elégtelen virilizációval járó DSD-k közül a leggyakoribb (2). 1953-ban J. M. Morris írta le a betegséget, mint testicularis feminizáció, a pontos genetikai hátteret csak később ismerték fel (5). Az X-kromoszómán található AR-génben bekövetkező mutáció révén androgénhatás nem alakul ki (1). Embrionális korban az Y-kromoszóma révén CAIS esetén is normál, funkcionáló herék fejlődnek. A herékben a Sertolli-sejtek anti-Müller-hormont (AMH) termelnek, amelynek hatására Müller-cső degradálódik, így belső női nemi szervek (uterus, proximális hüvely) nem fejlődnek ki (4, 5). Ugyanakkor a Leydig-sejtek által termelt tesztoszteron az AR mutációjának köszönhetően nem képes a férfi belső, illetve külső nemi szervek fejlődését indukálni. A külső nemi szerveik nőiesek, a hüvely rövidebb (2-8 cm), vakon végződik (4). Pubertás során a zsírszövetben végbemenő aromatáz enzim által katalizált tesztoszteron-ösztrogén átalakulás révén nőies másodlagos nemi jellegek alakulnak ki. A nemi szőrzet ritka vagy teljesen hiányzik (4, 5, 6). A pszichoszociális nem általában szintén női (4).
Gyermekkorban az első figyelemfelhívó tünet, amivel orvoshoz fordulnak, a leánygyermekeknél jelentkező lágyéksérv (7). Deeb és munkatársai által végzett felmérésből kiderül, hogy az Egyesült Királyságban a CAIS-betegek 57%-ának volt inguinalis sérve az első megjelenéskor (8). A gyermekkori sérvek előfordulási gyakorisága 1-4%, továbbá sokkal gyakrabban jelentkeznek fiúgyermekekben (10:1) (4). Az előbbiek miatt leányoknál jelentkező lágyéksérv esetén a kariotipizálás és további kivizsgálás ajánlott lehet a félrekezelések elkerülése érdekében (8). A pubertás elindulását követően a második leggyakoribb ok, ami a kivizsgálást elindítja, a primer amenorrhoea, ahogyan esetünkben is láthattuk. Ilyenkor gyakran összetéveszthető az MRKH-szindrómával, mivel a klinikai megjelenés hasonló. Az MRKH-szindróma vagy Müller-cső-aplázia a méh és a hüvely proximális 1/3-ának fejlődésének elmaradásával járó rendellenesség, azonban a kariotípus ebben az esetben 46, XX. Esetünkben a kivizsgálás első szakaszában is a CAIS-nál gyakrabban előforduló MRKH-szindróma merült fel (1:5000 leány születés) (9). A korai CAIS-diagnózist napjainkban elősegítheti a prenatális genetikai diagnosztika elterjedése. Az anyai vérből vett magzati DNS-fragmentumok alapján végzett genetikai vizsgálatok során a pontos kariotípus megállapítható, így amennyiben az eltér az ultrahangvizsgálat során vagy születéskor látható fenotípustól a CAIS gyanúja felmerülhet (4).
Az androgén inszenzitivitásnak ismert részleges formája is, az úgy nevezett parciális androgén inszenzitivitási szindróma (PAIS). A klinikai megjelenés ezeknél az eseteknél változatos attól függően, hogy milyen mértékben van AR funkció, és androgén hatás. Atípusos férfi külső genitáliák jellemzőek, enyhébb formáknál hypospadiasis, súlyosabb esetben micropenis figyelhető meg. Pubertáskorban gyakori a gynecomastia. A pszichoszociális nem általában férfi. (5) A legenyhébb forma MAIS (mild androgen insensitivity syndrome), ahol jellemző az AR mutáció, de nincs külső genitáliákban eltérés. Általában csak későn, meddőségi kivizsgálás kapcsán fedezik fel.
A CAIS korai diagnózisa azért fontos, mert a kor előrehaladtával a hasüregben, lágyékcsatornában elhelyezkedő herében a csírasejtes in situ neoplázia (GCNIS) és a malignus heredaganat megjelenése növekszik. Dean és munkatársai az irodalmi adatokat áttekintve azt találta, hogy a CAIS miatt végzett profilaktikus gonadectomiákat követően a heredaganatok incidenciája gyermekkorban 0,8-2% volt, míg felnőttkorban 16% (10). Továbbá a pubertás beindításában nagy szerepe van a gonádok hormontermelésének, ezért, amennyiben gyermekkorban történik a gonádok eltávolítása, a hormonpótlás pubertáskorban való elindítása kiemelten fontos (1, 5). PAIS esetén, mivel a gonádokban vannak működő csírasejtek a malignitás esélye magasabb, mint CAIS esetén, kb. 15%, de a kezeletlen esetekben akár 50% is lehet (5, 11). A gonadectomia szükségessége, időzítése a fentiek miatt nem pontosan tisztázott, a prepubertásban elvégzett műtét az alacsony malignitásrizikó miatt megfontolandó, azonban a nemzetközi irodalomban erre vonatkozó ajánlások nem állnak rendelkezése (10). A pubertás spontán megindulása miatt a gonadectomia halasztása mérlegelendő (12).
Esetünkben a beteg kora miatt a gonádok eltávolítását javasoltuk, amelyek rosszindulatú daganatot nem igazoltak. A herék lokalizációjának megfelelően végezhetünk inguinalis vagy laparoszkópos műtétet egyaránt, azonban még a minimál invazív technikáknál sem elhanyagolhatók a műtétből adódó lehetséges szövődmények, így a profilaktikus műtét elvégzése előtt a beteggel részletes konzultáció szükséges. PAIS esetén leánygyermekekben a pubertáskori virilizáció megelőzése és a magas malignitásrizikó miatt gonadectomia javasolt prepubertásban. Fiú PAIS-ban az orchidopexia az elsődleges kezelés, rendszeres utánkövetés mellett. (5) Amennyiben a beteg mégis a műtét halasztását választja, rendszeres képalkotó vizsgálatok (UH, MR) (11), tumormarker-vizsgálatok (AFP, b-hCG, LDH) és szükséges esetén biopszia végzendő, de jelenleg egyértelmű ajánlások nincsenek a pontos követési stratégiához (5). A műtétet követően fertilis korban is szükséges a hormonpótlás a másodlagos nemi jellegek, megfelelő csontsűrűség megtartása érdekében a fiziológiás menopauzáig (7). A hormonpótlás leggyakrabban ösztradiollal történik, amely adható per os vagy transzdermális formában is. Léteznek vizsgálatok a transzdermálisan adott tesztoszteronnal megvalósított hormonpótlásra is. Birnbaum és munkatársai randomizált, kettős vak vizsgálatban összehasonlították a transzdermálisan alkalmazott tesztoszteron és ösztrogén hatásait. Eredményeik alapján a két kezelés egyformán hatékony mind a hormonszintek, mind a pszichoszociális tényezőket tekintve. Bár a tesztoszteroncsoportban több mellkékhatást észleltek mindkét kezelés jól tolerálható volt. A tesztoszteron esetében virilizáció nem jelentkezett, tovább ebben a csoportban jobb szexuális funkcióról számoltak be a betegek (13). Hormonkezelés mellett a rendszeres endokrinológiai, csontdenzitás-vizsgálatok szintén elengedhetetlenek. A nemi élet javítása céljából hüvelyi dilatátorok használata, azok sikertelensége esetén rekonstrukciós beavatkozások is szükségessé válhatnak (7).
Következtetések
A CAIS egy ritka genetikai rendellenesség női fenotípusú egyének között, amely gyakran későn kerül felismerésre. A korai diagnózis a magasabb malignitásrizikó miatt fontos lehet. A CAIS gyanúja akár már prenatális korban is felmerülhet, de típusos esetekben, gyermekkorban is diagnosztizálható. A férfi retineált gonádokban nagyobb eséllyel alakulhatnak ki
rosszindulatú daganatok, amelyek előfordulása irodalmi adatok alapján úgy tűnik, hogy a korral gyakoribbá válik. Egyértelmű ajánlások ugyan nem állnak rendelkezésre, de a profilaktikus gonadectomia a pubertás utáni időszakra halasztható a spontán pubertás lezajlása érdekében. Felnőttkorban a megelőző műtét javasolandó, de a döntést a beteggel egyetértésben kell meghozni. A műtétet követően hormonpótlás szükséges rendszeres követés mellett.
Irodalom
https://doi.org/10.6065/apem.2040170.085
2. Walia R, Singla M, Vaiphei K, et al. Disorders of sex development: a study of 194 cases. Endocr Connect 2018 Feb; 7(2): 364–371.
https://doi.org/10.1530/EC-18-0022
3. Gillingham AW, Kenton K, Geynisman-Tan J, et al. Laparoscopic Vecchietti – Minimally Invasive Treatment for Vaginal Agenesis, Journal of Minimally Invasive Gynecology 2019; 26(7): Supplement
https://doi.org/10.1016/j.jmig.2019.09.605
4. Tyutyusheva N, Mancini I, Baroncelli GI, et al. Complete Androgen Insensitivity Syndrome: From Bench to Bed. Int J Mol Sci 2021 Jan 27; 22(3): 1264. https://doi.org/10.3390/ijms22031264
5. Batista RL, Costa EMF, Rodrigues AS, et al. Androgen insensitivity syndrome: a review. Arch Endocrinol Metab 2018 Mar-Apr; 62(2): 227–235. https://doi.org/10.20945/2359-3997000000031
6. Gulía C, Baldassarra S, Zangari A, et al. Androgen insensitivity syndrome. Eur Rev Med Pharmacol Sci 2018 Jun; 22(12): 3873–3887.
https://doi.org/10.26355/eurrev_201806_15272
7. Guo M, Huang JC, Li CF, et al. Complete androgen insensitivity syndrome: a case report and literature review. J Int Med Res 2023 Feb; 51(2) https://doi.org/10.1177/03000605231154413
8. Deeb A, Hughes IA. Inguinal hernia in female infants: A cue to check the sex chromosomes? BJU Int 2005; 96: 401–403.
https://doi.org/10.1111/j.1464-410X.2005.05639
9. Herlin MK, Petersen MB, Brännström M. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: a comprehensive update. Orphanet J Rare Dis 2020; 15: 214. https://doi.org/10.1186/s13023-020-01491-9
10. Deans R, Creighton SM, Liao LM, et al. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): patient preferences and clinical evidence. Clinical Endocrinology 2012; 76: 894–898. https://doi.org/10.1111/j.1365-2265.2012.04330.x
11. Döhnert U, Wünsch L, Hiort O. Gonadectomy in Complete Androgen Insensitivity Syndrome: Why and When? Sex Dev 2017; 11(4): 171–174. https://doi.org/10.1159/000478082
12. Birnbaum W, Marshall L, Werner R, et al. Oestrogen versus androgen in hormone-replacement therapy for complete androgen insensitivity syndrome: a multicentre, randomised, double-dummy, double-blind crossover trial. Lancet Diabetes Endocrinol 2018 Oct; 6 (10): 771–78.
https://doi.org/10.1016/S2213-8587(18)30197-9